



























Cuando el coronavirus pandémico SARS-CoV-2 causante de la Covid-19 apareció en la escena pública de forma oficial (diciembre de 2020), ya acarreaba mutaciones (lo lleva escrito en su genoma de casi 30.000 letras). Según la OMS, el origen zoonótico está en alguna especie de murciélago de herradura (Rhinolophus spp.). Se desconoce si, en el viaje evolutivo desde el mamífero volador al ser humano, el coronavirus pasó antes por uno o varios animales intermediarios. En tal supuesto, habría un aumento de nuevos ajustes adaptativos. El SARS-CoV-2, como virus RNA, surgió, llegó y sigue mutando.
Son muchas las mutaciones (cambios o sustituciones) de aminoácidos, en todos los segmentos del largo genoma viral. También hay deleciones (pérdidas de algunos aminoácidos). Probablemente, todos los cambios son importantes, aunque, como en la granja en rebelión de Georges Orwell, unos son más que otros. Las mutaciones y deleciones en la proteína S, Spike o espiga parecen ser las de mayor importancia epidemiológica (contagios), patológica (gravedad) y terapéutica (peor respuesta a los tratamientos con anticuerpos monoclonales). También preventiva (menor eficacia de las vacunas).
Desde el punto de vista estructural, la espiga es una proteína trimérica (tres componentes o monómeros) de forma parecida a un segmento de brócoli o coliflor. Es necesaria para que el virus se una al receptor de la membrana plasmática celular (ACE2) antes de entrar en las células. El asunto es bastante más complejo (por ejemplo, la necesaria participación de algún correceptor y de la furina), pero suficiente por ahora. La espiga, además, es un antígeno con capacidad de generar respuesta de anticuerpos.
Las mutaciones son muy numerosas. Determinadas combinaciones de ellas originan diversas variantes de preocupación y de interés, como expusimos en una colaboración anterior. Algunas destacan por cambiar la funcionalidad del virus. Su importancia, hasta donde sabemos, es muy diferente. Destacamos aquí las que parecen más notables.
Un consorcio de mutaciones
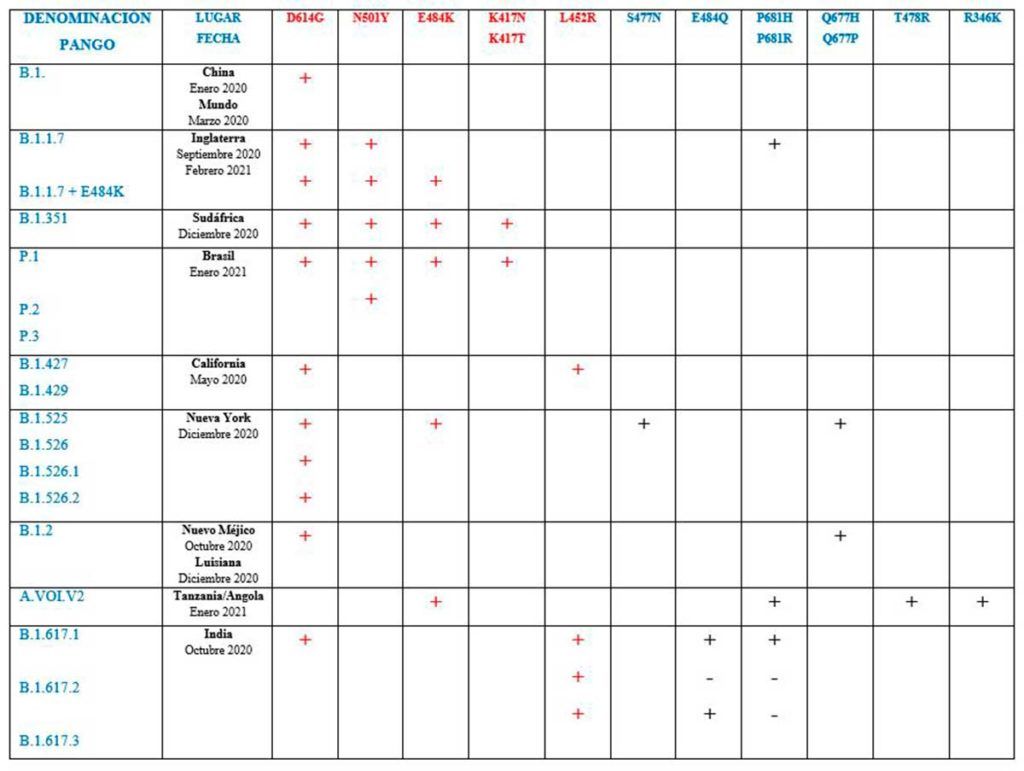
Seleccionamos solo cinco mutaciones de importancia. Están presentes en las cuatro variantes de preocupación (variants of concern/VOC) y participan en algunas variantes de interés (variants of interest/VOI). Hasta el momento, no hay ninguna que suponga una variante de alta consecuencia (CDC: Variant of High Consequence/VOHC) frente a las que fracasan las medidas no farmacológicas.
Estas mutaciones, cuando se agrupan en un virus concreto, conforman un consorcio de mutaciones (Tabla 1). Un consorcio, según la primera de las tres acepciones de la palabra en el diccionario de la RAE, es una asociación de empresas o entidades con intereses comunes para participar conjuntamente en un proyecto o negocio importante. Naturalmente aquí, las empresas o entidades son las variantes virales mutantes. Los intereses comunes estriban en dominar sobre otras variantes virales que carecen de tales cambios genéticos. Es pura evolución.
Mutación D614G. Infectividad: + / Virulencia: – / Respuesta a anticuerpos: +
Al poco de que el virus original (WU-Hu-1) empezara a arrasar a la humanidad, apareció la mutación denominada D614G. El cambio de un solo aminoácido (ácido aspártico o D por glicina o G) sirve al virus para unirse mejor al receptor ACE2 celular. Es el inicio del proceso que el virus necesita para reproducirse en miles de copias nuevas. Un fenómeno biológico en el que se comenten errores por azar. Los errores son nuevas mutaciones. La mayoría no van a más. Algunos hacen al virus más eficaz entre sus congéneres y le permiten hacerse dominante. Ocurre como en la champions futbolera. Aquí es la Darwin’s league. En virtud de esto, la mutación D614G se hizo dominante en dos o tres meses. Ocupó las estadísticas mundiales en un 90%. Así durante casi todo el año 2020. Como curiosidad, parece ligada a la pérdida del olfato (anosmia) sufrida por muchos pacientes.
Mutación E484K. Infectividad: + / Virulencia: ¿? / Respuesta a anticuerpos: –
En Sudáfrica se detectó, en mayo de 2020, la variante B.1.351. Su mutación estrella es la K484N. Un cambio de glutámico (K) a lisina (N). Permite a los virus eludir, en distinto grado, los anticuerpos naturales y vacunales que pretenden neutralizarlos. Es una mutación de escape. La variante B.1.351 también tiene las mutaciones D614G y N501Y. Por tanto, a su capacidad elusiva de la inmunidad suma la mayor difusión. Es más transmisible (50%) y no se sabe si causa mayor gravedad.
En Manaos (Brasil), en noviembre de 2020, se detectó una variante muy parecida a la B.1.351 porque tiene las mutaciones D614G, N501Y y E484K. Se denomina P.1. Solo dos meses más tarde fue detectada en el aeropuerto de Tokio (Japón) en una familia de cuatro miembros regresados de Manaos (Brasil). Hoy se conocen dos subvariantes más de la P.1. Se las llama P.2 y P.3. La variante P.1., como su pariente la B.1.351, puede ser más transmisible, no se sabe si más grave y también se desconoce la eficacia de las vacunas.
La mutación E484K también ha sido detectada en algunos genomas de la variante B.1.1.7. Se la conoce como B.1.1.7 con E484K.
Mutación N501Y. Infectividad: + / Virulencia: ¿? / Respuesta a anticuerpos: +
En septiembre de 2020 se detectó una nueva variante en el condado de Kent (suroeste de Inglaterra). Se denomina B.1.1.7 (la prensa y muchos comunicadores la llaman variante británica). Se caracteriza por presentar la mutación N501Y en la espiga, originada al cambiar el aminoácido asparagina (N) por tirosina (Y), además de la D614G y otras cinco más y dos deleciones (69/70 y 144/144). Se caracteriza funcionalmente por permitir al virus una mejor y más intensa unión al receptor celular ACE2.
Como rasgo epidemiológico tiene una mayor capacidad de contagio (50%), es posible que cause enfermedad más grave, aunque las vacunas parecen todavía efectivas. Hoy (mayo de 2021), la B.1.1.7 es la variante que domina en el mundo.
Mutación L452R. Infectividad: + / Virulencia: + / Respuesta a anticuerpos: + (¿menos a linfocitos T?)
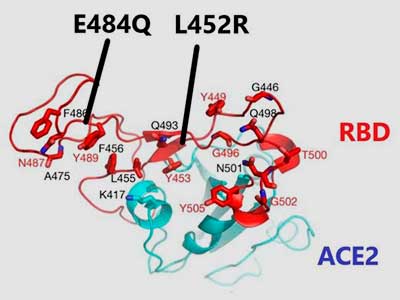
En California, sobre todo en los Ángeles, acabaron el año 2020 con un grave problema de incremento de casos. Los estudios de secuenciación genómica permitieron detectar en septiembre una mutación del aminoácido leucina (L) por arginina (R). La mutación L452R hace a la variante B.1.427/B.1.429 más transmisible (20%), puede ser que cause mayor gravedad y parece responder a las vacunas. Existen sospechas de una posible resistencia a las células T. Pero esta variante parece estar en decadencia (como ocurrió en marzo de 2020 con el denominado Clúster 5 danés asociado a los visones).
La mutación L452R también es clave, junto con otras dos mutaciones (E484Q y P681H/R), (Figura 1) en la variante o linaje ahora de moda: B.1.617.1, mal llamada India y una subvariante de la misma (B.1.617.2). Es interesante resaltar que E484K y E484Q son muy parecidas pero distintas.
En la primera el ácido glutámico (E) es sustituido por lisina (K) y en la segunda por glutamina (Q). Ambas reducen la respuesta de anticuerpos por un factor 10 y 6, respectivamente. Lo interesante, según el Dr. Ravi Gupta, de Cambridge, es que la asociación E484Q y L452R (mal llamada doble mutante) lo reduce por cuatro.
En un estudio realizado en Israel, aunque con pocos casos, la respuesta a la vacuna mRNA de Pfizer contra esta variante B.1.617 alcanza el 98% de eficacia.
Mutación K417N/T. Infectividad: + / Virulencia: ¿? / Respuesta a anticuerpos: –
Se caracteriza por el cambio del aminoácido lisina (K) por asparagina (N) o treonina (T). Es una mutación presente en dos de las variantes o linajes de preocupación: B.1.351 y P.1. Por tanto, suma a sus cualidades la capacidad de unión más intensa al receptor celular ACE2.
Comentario
En esta breve reseña sobre mutaciones del SARS-CoV-2 no están todas las que son (son muchas, y cada vez más) pero, en lo referido a las variantes de preocupación, son todas las que están. El fenómeno mutante es dinámico y se activa a medida que aumenta el número de contagios. Parece que la evolución convergente es el proceso biológico que subyace en tan complejo como azaroso asunto. El virus hace su trabajo.
En las conductas y comportamientos humanos quizás resida la clave de la aparición y rápida difusión de los linajes mutantes. Desde California a Nueva York, desde Inglaterra a España, desde Italia a Francia o desde Brasil a Argentina, las conductas inadecuadas han ido y van a favor de la propagación del virus. Esta opinión que personalmente defendemos desde el comienzo de la pandemia, la refrenda un estudio reciente de Science Translational Medicine en el estado de Washington.
Lo que está sucediendo ahora en India es el ejemplo más dramático de la situación. No parece, por ahora, que la variante B.1.617 sea la culpable del desastre asiático. O, al menos, no es la única causa. A medida que se van haciendo más estudios genómicos en todo el mundo, habrá más información sobre los virus mutantes. Mientras llegan, parece razonable pensar que, donde aparecen las variantes que preocupan e interesan, además de viajeros portadores hay un descontrol epidemiológico de la pandemia.
El consorcio de mutaciones, definitorias de algunas variantes, solo está advirtiendo de que, muy probablemente, los humanos no estamos haciendo las cosas como deberíamos. Ni aún con las vacunas vigentes, cuyo ritmo de ejecución y amplitud geográfica de distribución es bastante menor del conveniente.
- Nota Este artículo y la tabla acompañante se basan en los datos de GISAID, Covariants.org, Nextstrain.org, Outbreak.info, PANGO lineages, la página Web del gobierno británico (GOV.UK), la página de los CDC, el trazador de variantes del periódico The New York Times y varios artículos científicos de la literatura internacional.







 importa")










