


El método de edición de ácido desoxirribonucleíco (ADN), la molécula de la vida, lo han utilizado en un modelo de ratón con progeria investigadores de varios centros estadounidenses, entre ellos el Instituto Nacional de Investigación del Genoma Humano (NHGRI), el Broad Institute de Harvard y el MIT, en Boston, así como el Centro Médico de la Universidad de Vanderbilt, en Tennessee.
El éxito de esta técnica de edición de bases de ADN es que han logrado corregir el trastorno del envejecimiento acelerado, como describen en Nature.


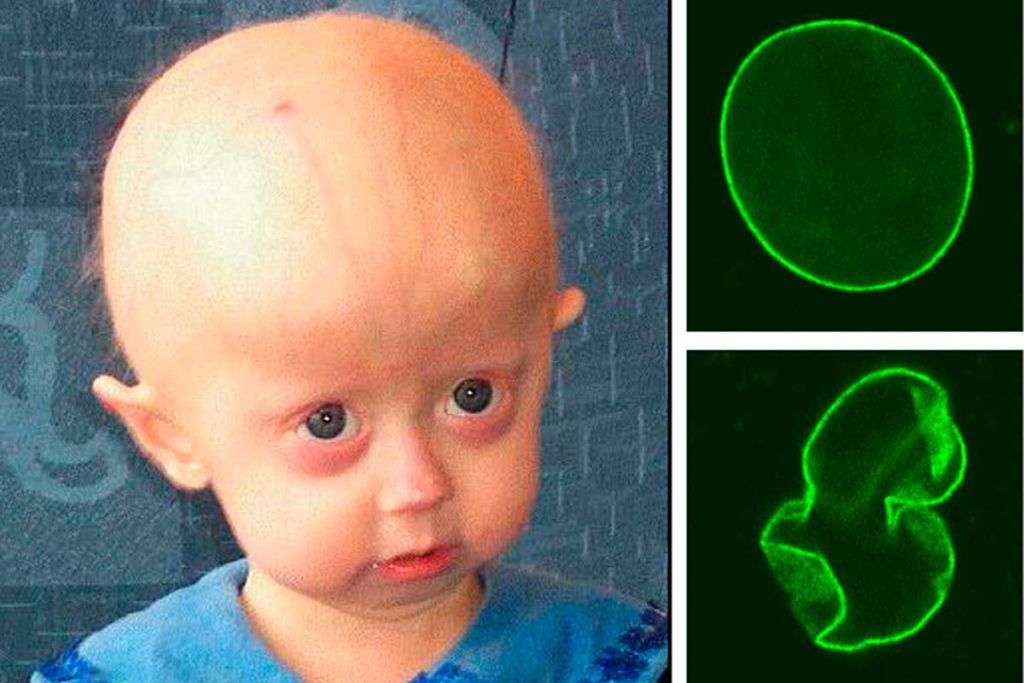
Esta enfermedad genética rara, también conocida como síndrome de Hutchinson-Gilford, se caracteriza porque causa un envejecimiento prematuro extremo en los niños y puede acortar significativamente su esperanza de vida.
Como se sabe, el ADN se compone de cuatro bases químicas: A, C, G y T. La progeria la causa una mutación en el gen nuclear LMNA, en el cual la base C se cambia a T. Este cambio aumenta la producción de la proteína tóxica progerina, que provoca el rápido proceso de envejecimiento.
Aproximadamente uno de cada cuatro millones de niños son diagnosticados con progeria en los dos primeros años de vida y prácticamente todos ellos desarrollan problemas de salud en la infancia y la adolescencia que, normalmente, están asociados con la vejez, incluidas las enfermedades cardiovasculares (ataques cardíacos y accidentes cerebrovasculares), pérdida de pelo, problemas esqueléticos, disminución de grasa subcutánea y piel endurecida.
Primer tratamiento terapéutico para la progeria
Para este estudio, los investigadores utilizaron una innovadora técnica de edición de ADN que se conoce como edición de base. Con ella se sustituye una sola letra de ADN por otra sin dañar, para estudiar cómo el cambio de esta mutación podría afectar los síntomas similares a la progeria en ratones.
El profesor Francis Collins, director de los Institutos Nacionales de Salud (NIH) de Estados Unidos y autor principal de este estudio, asegura que “no se puede subestimar el costo de esta devastadora enfermedad en los niños y sus familias. El hecho de que una sola mutación específica cause la enfermedad en casi todos los niños afectados nos hizo darnos cuenta de que podríamos tener herramientas para encontrar una solución. Estas herramientas solo podrían desarrollarse gracias a inversiones a largo plazo en la investigación básica de la genómica”.
El estudio sigue otro hito reciente para la investigación de la progeria, ya que la FDA aprobó el primer tratamiento terapéutico para la progeria en noviembre pasado. Se trata del fármaco Lonafarnib.
Es un inhibidor de la farnesiltransferasa (FTI). Hasta ese momento no había opciones que demostrasen la prolongación de vida de los niños afectados, cuya media de supervivencia es de 14 años.
En el Hospital Infantil de Boston se llevó a cabo un estudio con 250 niños de distintos continentes. En las conclusiones se demostró que había una relación entre Lonafarnib y el aumento de la supervivencia. Este fármaco, desarrollado por científicos de la multinacional Merck Sharp & Dohme, inhibe la enzima farnesiltranserasa e impide así que la proteína mutada pase a la pared celular, que es donde está gran parte del daño.
Técnica de edición de bases de ADN
Aunque la terapia con medicamentos proporciona cierta extensión de vida, no es una cura de la enfermedad, por lo que el método de edición de ADN puede proporcionar una opción de tratamiento adicional en un futuro que cada vez está más próximo.
Para David Liu, del Broad Institute donde desarrollaron el método de edición de base en 2016, financiado en parte por NHGRI, la edición CRISPR, aunque revolucionaria, todavía no puede realizar cambios precisos en el ADN en muchos tipos de células.
“La técnica de edición de bases que hemos desarrollado -explica- es como una función de buscar y reemplazar en un procesador de texto. Es extremadamente eficiente para convertir un par de bases en otro, lo que pensamos que sería eficaz para tratar una enfermedad como la progeria”.
Para probar la efectividad de su método de edición de bases, el equipo colaboró inicialmente con la Fundación de Investigación de Progeria para obtener células de tejido conectivo de pacientes con esta enfermedad ultrarara. El tratamiento fijó la mutación en el 90% de las células.
Doblaron la esperanza de vida en roedores con progeria
Tras este éxito, los investigadores probaron la técnica de edición de genes administrando una única inyección intravenosa de la mezcla de edición de ADN en casi una docena de ratones con la mutación que causa la progeria poco después del nacimiento. El editor de genes restauró con éxito la secuencia de ADN normal del gen LMNA. Y lo consiguió en un porcentaje significativo de células en varios órganos, entre ellos el corazón y la aorta.
Muchos de los tipos de células de los ratones aún mantenían la secuencia de ADN corregida seis meses después del tratamiento. En la aorta, los resultados fueron incluso mejores de lo esperado, ya que las células editadas parecían haber reemplazado a las que portaban la mutación de progeria.
Lo más interesante es que la esperanza de vida de los ratones tratados aumentó de siete meses a casi 1,5 años. La esperanza de vida normal promedio de los ratones utilizados en el estudio es de dos años.
Por último y en opinión del profesor Jonathan D. Brown, del Centro Médico de la Universidad de Vanderbilt, “en última instancia, nuestro objetivo será intentar desarrollar esto para las personas, pero hay preguntas clave adicionales que debemos abordar primero en estos sistemas modelo».